















 75 148
75 148

co-occurred, while
FH/TERT
(
p
= 0.04) were mutually
exclusive.
3.3.
Comparison to the TCGA data set
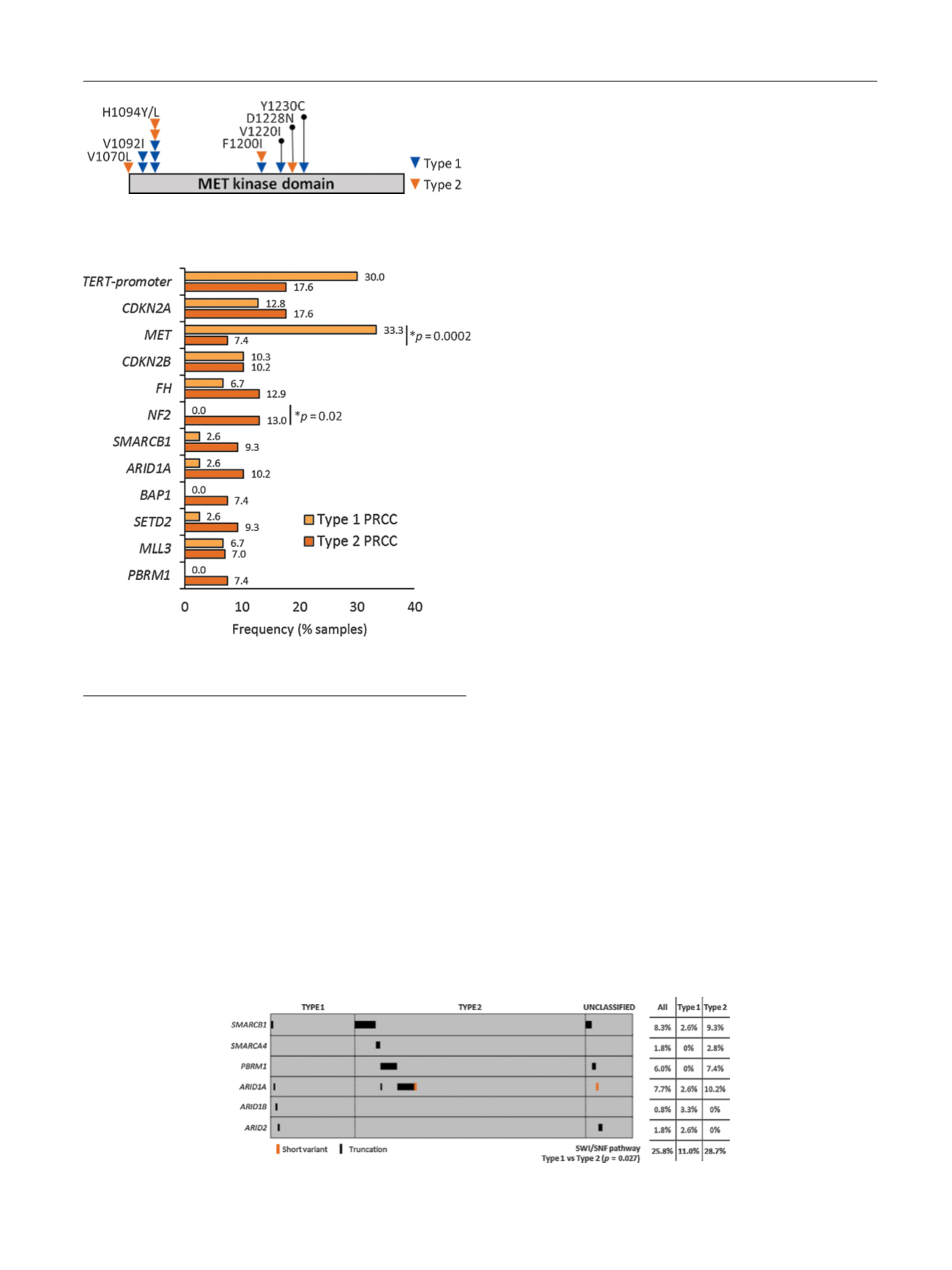
Fig. 5highlights differences in GA frequency in our data set
and the TCGA cohort. In both type 1 and type 2 disease, the
frequency of
MET
alterations (short variant mutations and
amplifications) is higher in our cohort (33% vs 17% for type
1; 7% vs 2% for type 2). By contrast,
MET
fusions were not
detected in this series but were observed in 1.3% of type
1 and 1.7% of type 2 cases in the TCGA cohort.
Similarly, alterations in
CDKN2A/B, FH
,
NF2
, and
SMARCB1
were identified more frequently in our data set. By contrast,
TCGA reported a higher frequency of alterations in
MLL2
and
SETD2
in type 2 disease.
4.
Discussion
To the best of our knowledge, our study reflects the largest
effort focused on genomic profiling of PRCC in a population
with more advanced disease. Our population is also slightly
larger than the TCGA PRCC cohort and includes a very
distinct stage distribution
[1]. In contrast to the 3% of
patients with confirmed stage IV disease noted in the TCGA
cohort, 60% of patients in our cohort had stage IV disease.
Furthermore, many patients had locally advanced (stage III)
PRCC (21%), and only a small minority (5%) had no
assignment of stage. It is possible that the more advanced
stage for patients in our cohort led to the marked
discordance in genomic findings highlighted in
Fig. 5, with
higher frequencies of alterations in
MET
,
CDKN2A/B
,
FH
, and
multiple SWI/SNF pathway elements. One caveat is that the
TCGA study also evaluated low-level copy gains for
chromosome 7 (including single copy gains) in their
analysis of
MET
alterations, and our comparative analysis
was limited to high-level amplifications, which may be a
more robust biomarker for responses to MET inhibitors on
the basis of studies in lung cancer
[18].
The findings of this study have potential therapeutic
implications, especially given the preponderance of ad-
vanced disease in this cohort. The higher frequency of
alterations in
MET
than previously recognized supports the
ongoing efforts to explore MET antagonists in prospective
clinical trials. Furthermore, cases with
MET
alterations with
concurrent
EGFR
or
KRAS
GAs should be carefully evaluated
for response to MET inhibitors, since it has been shown that
EGFR/KRAS
alterations mediate resistance to MET inhibition
[19,20], although a patient with carcinoma of unknown
primary harboring
MET
amplification and
KRAS
GAs
responded to crizotinib
[21].
The dual VEGFR2/MET inhibitor foretinib has shown
impressive activity in metastatic PRCC, with PFS of 9.3 mo
and a particularly high response rate (50%) among patients
with germline
MET
mutation
[22]. Results from the EORTC
CREATE trial assessing crizotinib showed a similar response
rate of 50% among patients with type 1 PRCC and
MET
mutation
[23]. Data from a phase 2 study of savolitinib, a
[(Fig._2)TD$FIG]
Fig. 2 – Sites of alteration in the
MET
protooncogene in patients with
type 1 and 2 papillary renal cell carcinoma.
[(Fig._3)TD$FIG]
Fig. 3 – Frequency of selected alterations in type 1 versus type
2 papillary renal cell carcinoma (PRCC).
[(Fig._4)TD$FIG]
Fig. 4 – SWI/SNF pathway analysis in type 1, type 2, and unclassified papillary renal cell carcinoma.
E U R O P E A N U R O L O G Y 7 3 ( 2 0 1 8 ) 7 1 – 7 8
75