















 76 148
76 148

more potent and selective MET inhibitor, were recently
presented
[24]. The study included 109 patients with
metastatic PRCC; PFS was significantly longer in patients
with
MET
alterations (6.2 vs 1.4 mo;
p
= 0.002).
The ongoing SWOG 1500 trial (NCT02761057) compares
the conventional strategy of VEGF inhibition with sunitinib
to MET inhibition (with either crizotinib, savolitinib, or
cabozantinib) in an unselected population of PRCC patients.
Another study (SAVOIR) is exploring the activity of
savolitinib versus control sunitinib in patients with MET-
driven PRCC (NCT03091192). Detailed efforts to character-
ize
MET
mutational status and copy number alterations
(CNAs) will accompany this effort. As diverse mechanisms
underlying
MET
dysregulation have now described, includ-
ing MET splice variants and intronic mutations
[25,26], it is
important to recognize what analyses were performed, as
the absence of alterations in CGP is not synonymous with
lack of an altered MET pathway.
Beyond
MET
, our data point towards other alterations
with potential therapeutic relevance. The cumulative
incidence of
CDKN2A/B
alterations was far higher in our
data set compared to the TCGA. CDK4/6 inhibitors may be
effective in patients with alterations in
CDKN2A
[27,28]. More than a quarter of our cohort also had
alterations along the SWI/SNF pathway. Kim et al
[29]demonstrated a dependence of SWI/SNF-mutated cancer
cell lines on EZH2-mediated stabilization of the PRC2
complex. Recent evidence also points towards the potential
efficacy of emerging EZH2 inhibitors in preclinical models
with SWI/SNF pathway alteration
[30,31]. The GAs seen in
the current series also point to patients predisposed to a
response to either VEGF- or mTOR-directed therapy. As one
example,
NF2
alterations were observed in 13% of the study
population; we have previously reported that this may
confer sensitivity to everolimus
[32]. In summary, the
alterations documented in our study could support (1) trials
exploring novel therapeutic targets (eg, MET, CDK4/6, or
EZH2) or (2) trials exploring mTOR- or VEGF-directed
agents in biomarker-selected populations with advanced
PRCC.
Our study also cites the mutational burden present in
PRCC, which is relatively low. There is an emerging
association between tumor burden and response to
immunotherapy in many tumor types; in RCC, these data
sets are relatively small
[33,34]. However, PD-1 inhibition
seems to have some encouraging activity in patients with
PRCC; understanding the correlation between tumor
burden and response in this unique cohort of patients
would be important
[35].
Other reports have offered genomic data for patients
with PRCC, but these data sets either focus on cohorts with
predominantly localized disease (eg, TCGA) or offer scant
details pertaining to clinical stage. For instance, Albiges and
colleagues
[36]reported genomic characterization of a large
PRCC cohort. In total, 220 specimens from patients with
PRCC were accessioned from the French RCC Network.
Notably absent, however, are data pertaining to clinical
stage in this cohort. Their findings are focused largely on
MET
expression and CNAs, both of which were elevated
among patients segregated by type 1 and type 2 disease.
Thus, the implications for MET inhibitors may be beyond
just MET mutations, and higher expression or MET CNAs
may constitute an MET-driven phenotype in the absence of
mutations.
Several limitations of our study should be acknowl-
edged. First, clinical outcomes data were not available for
the majority of patients in our series. However, for a select
number of patients, exceptional responses (derived from
therapies matched to genomic profile) have been docu-
mented. For instance, one patient harboring an alteration
in
MET
(H1094L) had a profound response to crizotinib
therapy
[37] .Prospective efforts such as the previously
described SWOG 1500 trial will hopefully validate such
observations. Second, we acknowledge that the majority
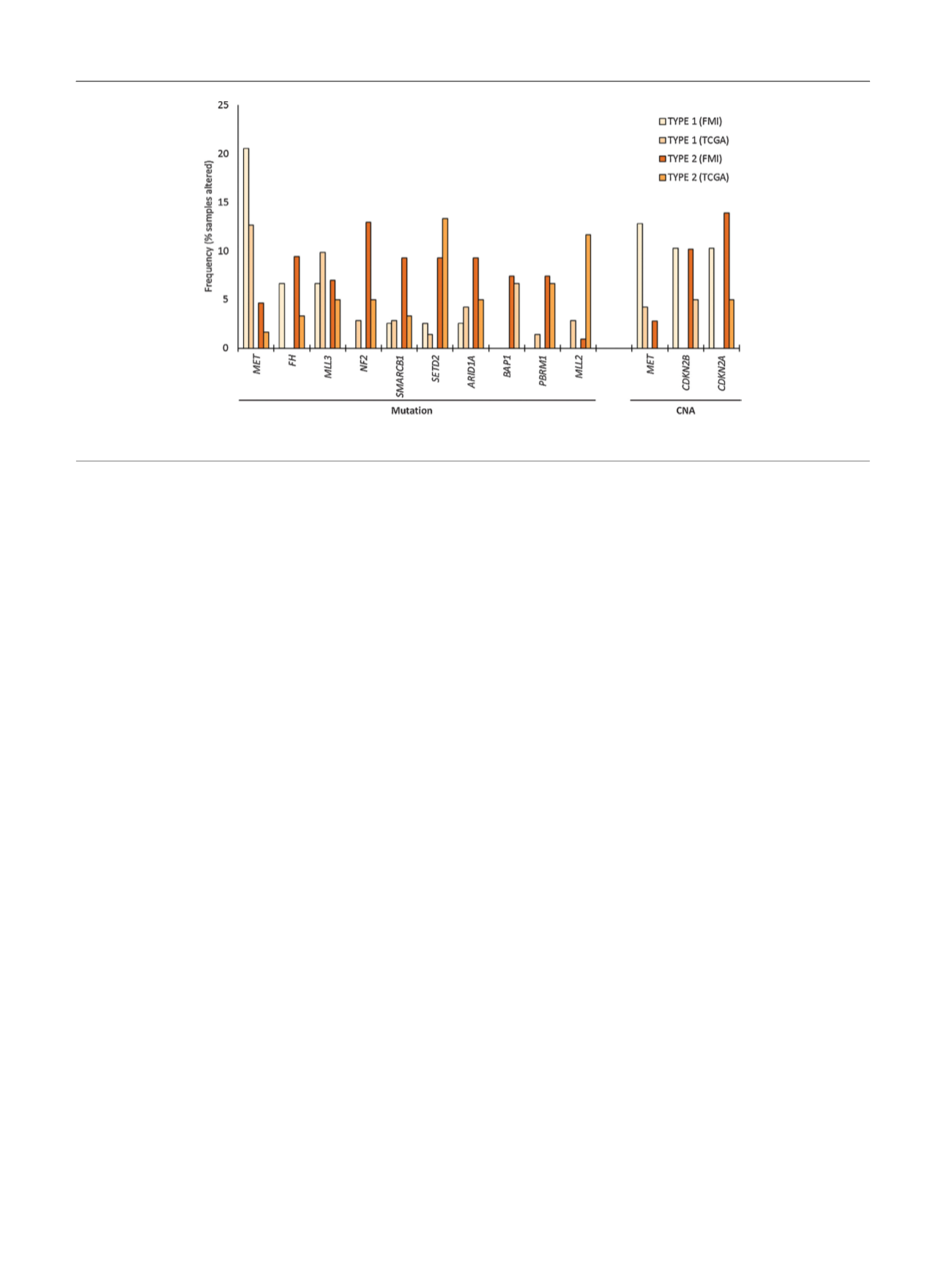
[(Fig._5)TD$FIG]
Fig. 5 – Frequency of genetic alterations in the current data set (FMI) versus The Cancer Genome Atlas (TCGA). CNA = copy number alteration.
E U R O P E A N U R O L O G Y 7 3 ( 2 0 1 8 ) 7 1 – 7 8
76